
Huan-Xiang Zhou
Professor and LAS Endowed Chair in the Natural Sciences
Contact
Address:
3462 SES
Office Phone:
Email:
CV Download:
Related Sites:
About Heading link
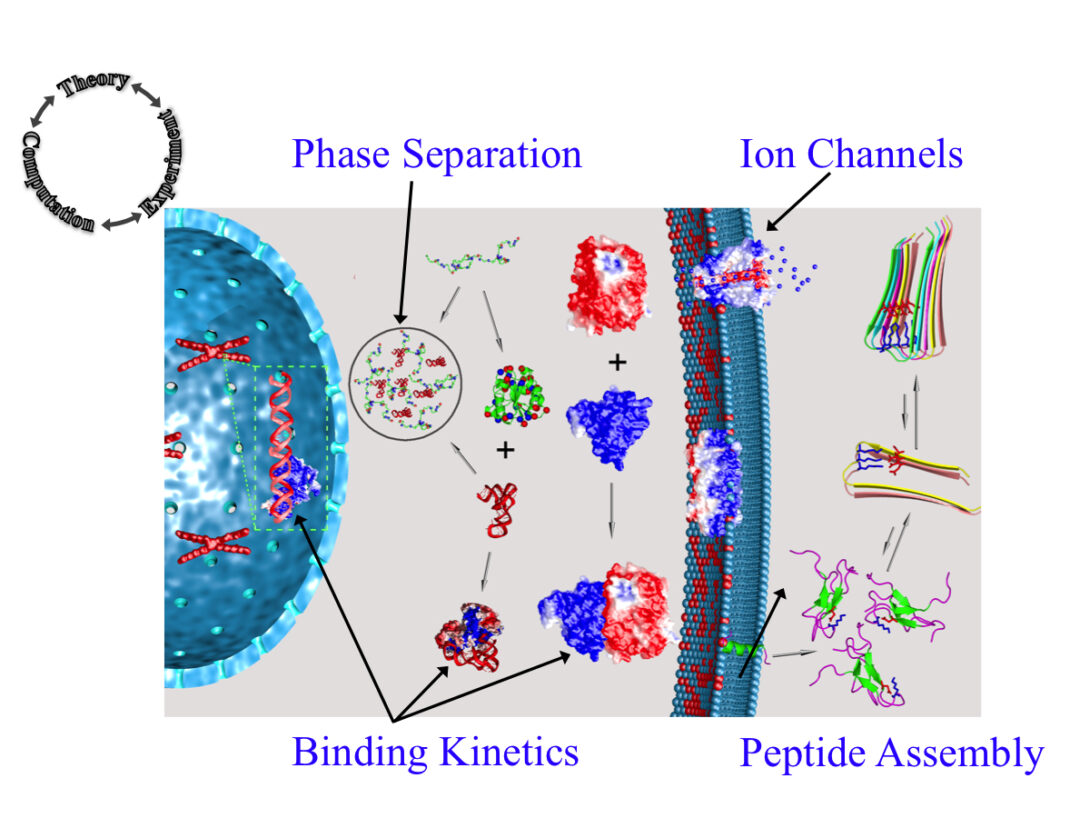
The Zhou group uses theory, computation, and experiment to address a range of topics in molecular and cellular biophysics. Four main areas of interest are: (1) liquid-liquid phase separation; (2) binding kinetics of intrinsically disordered proteins; (3) functional mechanisms of ion channels and other membrane proteins; and (4) self-assembly of peptides.
Liquid-Liquid Phase Separation
In cellular environments, proteins function in a milieu crowded with macromolecules. Macromolecular crowding can have profound effects on the conformational, thermodynamic, and kinetic properties of proteins. These effects are particularly strong for intrinsically disordered proteins (2018 BJ). Crowding can also lead to emergent behaviors, in particular liquid-liquid phase separation, by which concentrated proteins form micro-sized droplets surrounded by dilute bulk solution. Protein droplets exist inside cells, where they are known as membraneless organelles and perform a wide range of biological functions, including RNA processing, ribosome biogenesis, and sequestration of mRNA and proteins. Liquid-liquid phase separation is now a main focus of our research, both computationally and experimentally.
Body Heading link
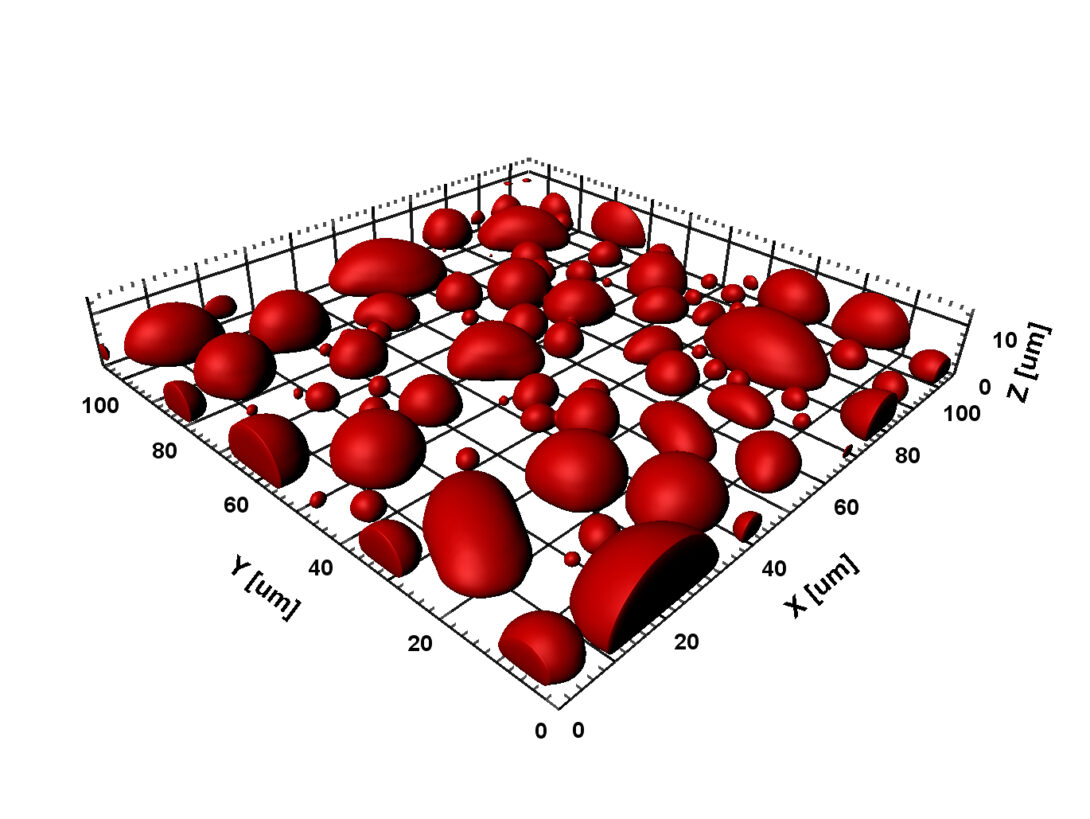
Our early work has helped spur the rapidly growing interest in macromolecular crowding. In 2009 we introduced a powerful approach for computing the effects of macromolecular crowding on the thermodynamic properties of proteins. This approach was implemented in 2013 and 2014 in a highly efficient manner via fast Fourier transform; hence the method is called FFT-based Modeling for Atomistic Protein-crowder interactions, or FMAP. In an exciting development, we have adapted this method to determine the phase boundaries of protein liquid-liquid phase separation (2016 JPCB). We are using FMAP and other methods to compute phase equilibria and integrating computation and experiment to elucidate the physical basis for the use of liquid-liquid phase separation in biological functions.
Binding Kinetics and Allostery
Proteins function by binding with small molecules, other proteins, nucleic acids, and macromolecular complexes. Biochemical processes quite often are under kinetic control, and therefore binding kinetics of proteins is of essential biological interest. Continuing a long-standing interest of the Zhou group, we are combining theoretical, computational, and experimental approaches to determine binding rate constants as well as binding mechanisms.
Body Heading link
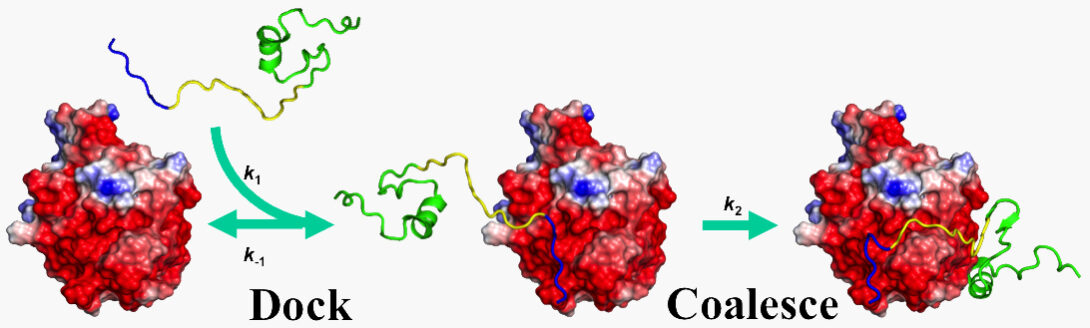
Since 1993, our theoretical and computational studies have contributed to a comprehensive physical understanding on the determinants for the magnitudes of binding rate constants and the determinants for binding pathways (2017 Annu Rev Biophys). A milestone was the development of an automated method for computing protein-protein binding rate constants (2011 Structure). We have since adapted this method, in combination with kinetic experiments, to elucidate the mechanisms for the binding of intrinsically disordered proteins to structured targets (2017 FEBS J). Current work aims to develop experimental methods for characterizing binding pathways of intrinsically disordered proteins and to design mutations to alter binding pathways.
Body Heading link
While some binding events directly lead to biological action, such as a chemical modification of the target molecule by an enzyme, other binding events serve to regulate the activities of proteins. The latter binding events occur at a site distant from the site of direct action, and this form of regulation is known as allostery. Since 2015, we have used molecular dynamics simulations to gain mechanistic insight on the allosteric regulation of a number of proteins (e.g., 2016 PNAS). We have particular interest in allosteric communications that rely not on large-scale conformational changes, but on changes in protein dynamics or conformational flexibility.
Body Heading link
Ion Channels and Other Membrane Proteins
Ion channels are proteins inserted into cell membranes to allow for passage of small ions. Using data from solid-state NMR and other structural techniques, we are calculating structures of ion channels and other membrane proteins inside membranes. These structures capture but a few snapshots of the proteins during their functional processes. Many other states can only be indirectly probed by experiments, notably ion current recordings on a whole cell or through a single channel. We are using computation, in particular molecular dynamics simulations, to provide structural models for states not accessible by experiments and to define pathways for the transitions between states.
Selected Publications
- A. Banks, S. Qin, K. L. Weiss, C. B. Stanley, and H.-X. Zhou (2018). Intrinsically disordered protein exhibits both compaction and expansion under macromolecular crowding. Biophys. J. 114, 1067-1079.
- L. Ou, M. Matthews, X. Pang, and H.-X. Zhou (2017). The dock-and-coalesce mechanism for the association of a WASP disordered region with the Cdc42 GTPase. FEBS. J. 284, 3381-3391.
- H.-X. Zhou (2017). Gating motions and stationary gating properties of ionotropic glutamate receptors: computation meets electrophysiology. Acc. Chem. Res. 50, 814-822.
- X. Pang and H.-X. Zhou (2017). Rate constants and mechanisms of protein-ligand binding. Annu. Rev. Biophys. 46, 105-130.
- C. Guo and H.-X. Zhou (2016). Unidirectional allostery in the regulatory subunit RIα facilitates efficient deactivation of protein kinase A. Proc. Natl. Acad. Sci. USA 113, E6776-E6785.
- S. Qin and H.-X. Zhou (2016). Fast method for computing chemical potentials and liquid-liquid phase equilibria of macromolecular solutions. J. Phys. Chem. B. 120, 8164-8174.
- A. R. Cormier, X. Pang, M. I. Zimmerman, H.-X. Zhou, and A. K. Paravastu (2013). Molecular structure of RADA16-I designer self-assembling peptide nanofibers. ACS Nano 7, 7562-7572.
- S. Qin, X. Pang, and H.-X. Zhou (2011). Automated prediction of protein association rate constants. Structure 19, 1744-1751.
- H. Dong and H.-X. Zhou (2011). Atomistic mechanism for the activation and desensitization of an AMPA-subtype glutamate receptor. Nat. Commun. 2, 354.
- M. Sharma, M. Yi, H. Dong, H. Qin, E. Peterson, D. D. Busath, H.-X. Zhou, and T. A. Cross (2010). Insight into the mechanism of the influenza A proton channel from a structure in a lipid bilayer. Science 330, 509-512.
Education
Ph.D., Drexel University, 1988