
Cynthia Jameson
Emeritus Professor
Contact
Address:
4240 SES, MC 111
Office Phone:
Email:
Related Sites:
APPLICATIONS OF XE NMR TO FUNDAMENTAL STUDIES OF ADSORPTION AND DIFFUSION IN NANO-CHANNELS, CAVITIES AND CAGES.
Jameson has contributed significantly to a fundamental understanding of the NMR chemical shift as a molecular electronic property. One characteristic of Jameson’s work is her interest in general trends and over-arching relationships, not just specific systems. Her contributions have usually involved providing the basis for a fundamental understanding of a limited body of data, and on this basis, predicting a systematic global pattern which emerges much later. The NMR chemical shift is a difference between two values of the nuclear magnetic shielding. Part of the motivation of Jameson’s work in NMR in the gas phase has been to understand the way in which intermolecular interactions affect the nuclear magnetic shielding, in other words, the exploration of the nuclear magnetic shielding surface, analogous to the intermolecular potential energy surface. In 1993 Jameson’s ab initio calculations showed that in the rare gas pair the shape of the purely theoretical shielding surface is indeed of a shape much like that predicted by Jameson nearly twenty years earlier. Almost all the reported studies of the effects of molecular collisions on the NMR chemical shift in the gas phase have emerged from Jameson’s laboratory. Her work forms the basis for the wide application of Xe NMR shifts for probing various polycrystalline surfaces, zeolites, other porous solids, and polymers.
Jameson’s studies of distribution and dynamic behavior of adsorbed species in what had been traditionally called “microporous” solids addresses a topical problem of both basic and technological importance. Actually the pores are in the nano-scale. What makes this research unusual is the combination of a detailed molecular level understanding of the fundamental processes with an awareness of the relevance of the results to potential technological applications. Jameson’s prior work focused on zeolite NaA, a well-characterized crystalline system. Jameson’s lab has determined directly the distribution of Xe atoms in the cavities of a microporous solid by observing individually the trapped clusters Xe, Xe2, Xe3, …, Xe8 in NMR. The chemical shifts of the clusters vary with temperature as they undergo changes in configurations within the cavities. For the first time Jameson reproduced the observed distributions, the cluster chemical shifts, and the temperature dependence by a computer simulation using only potential energy and shielding surfaces, with no adjustable parameters. These are the most detailed tests yet of any computer simulation. The observed quantities depend on the equilibrium distribution of the Xe between the bulk gas and inside the zeolite (the adsorption isotherm), the distribution of the adsorbed Xe among the cages (the cluster sizes and their fractions) and the distribution of the atoms of a given cluster within a cage (the chemical shift of the cluster depends on this). Her simulations provide these quantities for pure Xe as well as for mixtures of Xe with another molecule, and are tested by the detailed experiments in the same lab. This judicious choice of system where individual clusters Xen are observable in the 129Xe NMR spectrum avoids having to interpret exchange-averaged spectra, where all the contributions are convoluted into a single chemical shift value. These investigations provide a basic understanding and the results highlights the subtleties and intricacies of the contributions of various intermolecular interactions to the Xe shielding function. The investigation of pore sizes, channel cross sections, cation substitutions and distributions are all important issues relevant to applications in catalysis and separation science. In another set of experiments and simulations, Jameson’s lab has determined the rate of cage-to-cage migration of xenon atoms in a zeolite. This work provides the most detailed picture yet of the process of diffusion within a microporous solid. Competitive adsorption is a very important issue in the technological applications of zeolites. Jameson’s experiments and simulations of binary mixtures in zeolites has provided a very detailed picture of competitive adsorption and diffusion as never before possible, right down to directly observing for the first time mixed clusters such as XeKr, Xe2Kr, … Xe6Kr. In other cases where the co-adsorbed molecules (such as Ar, CO, CH4, CO2) are in fast exchange, she has directly determined the average number of co-adsorbed molecules in the same cage as exactly n Xe atoms for a given loading of the two sorbates. This level of detail is unprecedented in the entire history of adsorption studies.
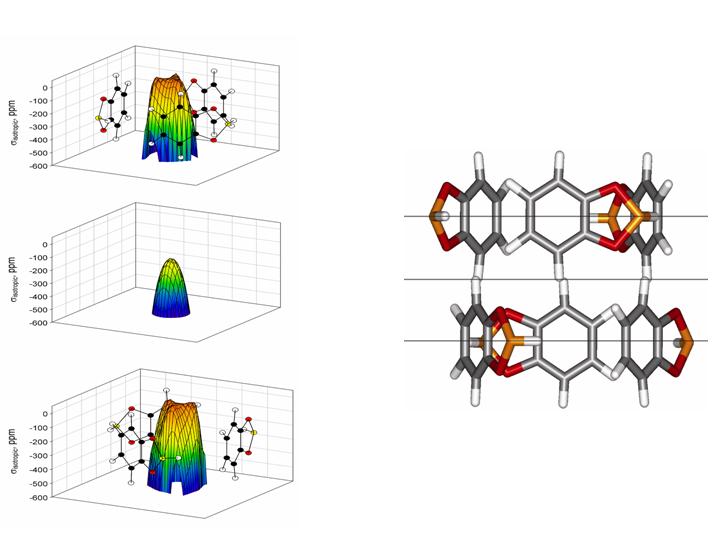
Simulations and observations of the average Xe chemical shift tensors for Xe in nanochannels reveal attributes of the channel, such as its cross section and eccentricity. Predictions of the Xe line shapes showing tensor components as a function of Xe occupancy and of temperature have preceded many observations in her laboratory as well as in others. A particularly interesting example is the prediction of the Xe NMR line shapes as a function of overhead Xe pressure for Xe in the nanochannels of molecular crystals of dipeptides of alanylvaline and valyl alanine, which turned out to provide not only adsorption isotherms but also predicted the detailed changes in line shapes with Xe occupancy for both molecular crystals before the spectra were observed. Xe shielding for Xe located at various positions in various planes in a nanochannel are calculated, in order to obtain chemical shift maps. One example of isotropic shielding maps is shown above.
STUDIES OF XE IN FLEXIBLE CAGES AND IN SOLUTION; OBSERVATION OF MOLECULAR TRANSPORT ACROSS MODEL MEMBRANES.
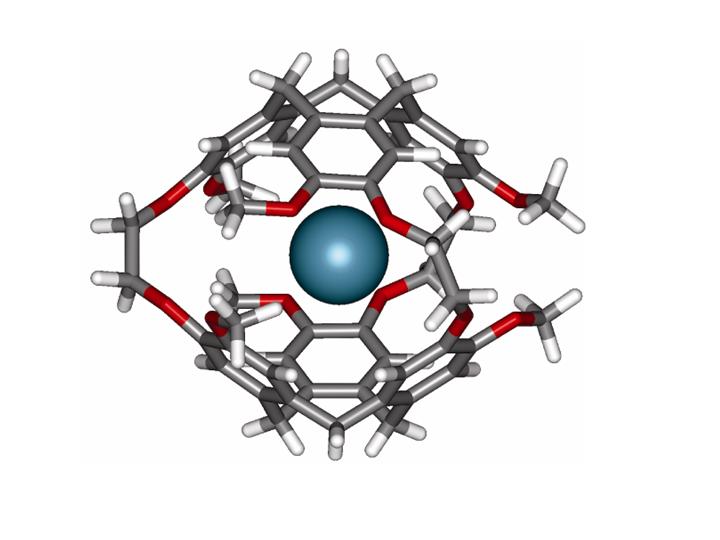
The determination of the mechanism of the sensing action of a biosensor, consisting of Xe in a flexible cage-shaped organic molecule (cryptophane-A) tethered to a molecular fragment which recognizes a specific protein, has been carried out by Jameson and Sears by starting with quantum calculations of of Xe shielding in the presence of the cage, and the simulations of Xe within the cage (See Xe in cryptophane cage above). Predictions of the Xe chemical shifts as a function of temperature, type of cage, and extent of deuteration of the cage matched experiments. Results of simulations were also consistent with SPINOE measures of average distances between Xe and various protons of the cage in various cages. Xe chemical shift simulations in a cage distorted away from the symmetrical structure of a free-floating cage (by being kept in a solution box with a protein) led to the prediction that the sensor shift relative to the case of no protein present, would be larger for shorter tethers and smaller for longer tethers, becoming zero for a tether long enough. Later experiments in the Pines lab proved this to be the case, thereby establishing the predicted mechanism for sensor action: i. e., the mechanical distortion of the symmetric cage by buffeting against the protein, as the sensor stays docked in the action of recognition, is detected by the Xe chemical shift since a cage slightly flattened against a protein has a smaller internal volume for the Xe, leading to closer interactions and larger chemical shifts.
Jameson’s predictions of chemical shifts of Xe in liquid solutions including water, alkanes, perfluoroalkanes and perfluorooctylbromide using molecular dynamics provided the first calculations of Xe shifts which for decades had been interpreted entirely in terms of refractive index of the solvent. The ability to reproduce the chemical shift of Xe in water is to be expected, since the shielding surfaces had previously been obtained by Jameson and Stueber from ab initio calculations for Xe shielding in various ice cages in clathrate hydrates, taking into account cage water molecules, water molecules in the next shell of hydrogen bonding partners, as well as electrostatic contributions from the remainder of other water molecules in the entire crystal. By doing molecular dynamics using a simulation box system introduced by S. Murad, not only solubilities and other average properties such as NMR chemical shifts in solutions can be obtained, but also rates of dynamic processes such as exchange, reorientation, permeation of channels, and ion transport in carbon nanotube models for ion channels.
Current studies of molecular transport across model membranes (with R. Gerald and S. Murad) involve experiments in a toroid cavity detector. The central Al rod is prepared with AAO pores to receive the transporting molecules. A model membrane is grown on the surface of the rod, electrostatically adhering to one end of a thiol that is anchored by an S-bond to the monolayer of gold sputtered onto the outer surface of the AAO. The lipid bilayer membrane is thus suspended over the honeycomb of pores. NMR imaging and spectroscopy in the concentric sections, from the center out to the cup containing the molecules in solution, will permit study of the transport process itself as the molecules are found successively at the model membrane surface, within the membrane, and through the membrane within the receiving pores on the other side. The richness of chemically specific information that can be obtained by doing various NMR spectroscopy experiments, besides NMR imaging, can provide details about the transport process not previously observed. This is in contrast to the typical studies of membrane transport by impedance measurements which count the ions that have made it across the membrane through the ion channel. Channel formers can be permitted to self-assemble under the proper conditions and insert into the model membrane to permit studies of transport through channels. The line shape signatures that indicate restricted motion at the membrane surface can be used to discover preferential alignment or other interactions of the molecule or the channel former with the membrane. In other words, the process of molecular transport itself can be investigated with this technique.
Selected Publications
- Measuring Chirality in NMR in the Presence of a Static Electric Field. Walls, J. D.; Harris, R.; Jameson, C. J. Chem. Phys. 2008, 128, 154502-1-6.
- Molecular Dynamics Simulations of Xe Chemical Shifts and Solubility in n-Alkanes. Yuan, H.; Murad, S.; Jameson, C.; Olson, J. J. Phys. Chem. C 2007, 111, 15771-15783.
- Diastereomeric Xe Chemical Shifts in Tethered Cryptophane Cages. Ruiz, E.; Sears, D.; Pines, A.; Jameson, C. J. Am. Chem. Soc. 2006, 128, 16980-16988.
- Ion Permeation Dynamics in Carbon Nanotubes. Liu, H. M.; Murad, S.; Jameson, C. J. Chem. Phys. 2006, 125, 084713-084713-14.
- Intermolecular Hyperfine Tensor for Xe@O2. Density and Temperature Dependence of Xe Chemical Shifts in Oxygen Gas. Vukovic, L.; Jameson, C.; Sears, D. Mol. Phys. 2006, 104, 1217-1225.
- Xe NMR Lineshapes in Channels of Peptide Molecular Crystals. Moudrakovski, I.; Soldatov, D.; Ripmeester, J.; Sears, D.; Jameson, C. P. Natl. Acad. Sci. U.S.A. 2004, 101, 17924-17929.
- Xe Chemical Shift Tensor in Silicalite and SSZ-24. Jameson, C. J. J. Am. Chem. Soc. 2004, 126, 10450-10456.
- The NMR Lineshapes of Xe in the Cages of Clathrate Hydrates. Jameson, C. J. and Stueber, D. J. Chem. Phys. 2004, 120, 10200-10214.
Education
BS, University of the Philippines, 1958
PhD, University of Illinois at Urbana-Champaign, 1963
Postdoctoral Fellow, Columbia University, 1963-64
Postdoctoral Fellow, University of Illinois at Urbana-Champaign, 1967
Visiting Scholar, Cambridge University, 1978, 1985, Oxford University, 1992-93, University of Milan-Bicocca, 2007
Program Officer for Experimental Physical Chemistry, National Science Foundation, 1990-91. University of Illinois Scholar, 1995-98
Miller Visiting Professor, University of California, Berkeley, 2002, AAAS Fellow since 1988